
Loss of Telomere Leads to p53 and Autophagy Induced Cell Death.
“autophagy-deficient cells … continued to proliferate and bypassed crisis” (2)
“In summary, cells in telomere crisis undergo cell death through autophagy, which is triggered by chromosome breakage and transduced by the cGAS–STING pathway. As cell death in crisis represents the final barrier against neoplastic transformation, a cancer therapy that involves inhibition of autophagy could be counterproductive… Moreover, cells lacking either cGAS or STING proliferated beyond crisis.“
“Autophagy mediates the turnover of cytoplasmic macromolecules to support cellular homeostasis. Autophagy generally blocks apoptosis, but in specific circumstances it can lead to cell death through excessive degradation of cell constituents. The authors studied telomere crisis using human fibroblasts and epithelial cells, in which the RB and/or p53 pathways were suppressed; these cells bypassed senescence and entered replicative stress, exhibiting telomere attrition, chromosome fusions and cell death.”
“Telomere deprotection through TRF2 depletion was sufficient to activate autophagy independently of replicative crisis, and genetic suppression of telomere fusions in TRF2-depleted cells reduced the accumulation of cytosolic DNA and attenuated autophagy, suggesting that fusion-dependent cytosolic DNA is required for the telomeric autophagy response.” (2)

“The cell fate of CPCs changes with age and is characterized by a switch away from proliferation and quiescence (reversible form of cell cycle arrest) towards senescence and increased basal commitment (‘irreversible’ forms of cell cycle arrest) accounting for age-associated stem cell exhaustion. Mechanistically, short telomeres activate p53 that induces autophagy and at least partially contributes to the age-associated change in cell fate. Blunting telomere shortening via overexpression of TERT-WT, silencing p53, or treating with pharmacological inhibitors of p53 (PFT) and autophagy (3-MA, Ulk1-In, BF) selectively attenuate senescence and basal commitment and reverse cell fate of aged CPCs.” (3)
Circadian Rhythm Controls Telomeres and Telomerase Activity.
• Telomerase and TERT mRNA expressions exhibit endogenous circadian rhythm.
• Human and mouse TERT mRNA expression are under the control of CLOCK–BMAL1 heterodimers.
• CLOCK deficient mice have shortened telomere length and abnormal oscillations of telomerase activity and TERT mRNA.
• Emergency physicians working in shifts lose the circadian rhythms of telomerase activity.
“Circadian clocks are fundamental machinery in organisms ranging from archaea to humans. Disruption of the circadian system is associated with premature aging in mice, but the molecular basis underlying this phenomenon is still unclear. In this study, we found that telomerase activity exhibits endogenous circadian rhythmicity in humans and mice. Human and mouse TERT mRNA expression oscillates with circadian rhythms and are under the control of CLOCK–BMAL1 heterodimers.
CLOCK deficiency in mice causes loss of rhythmic telomerase activities, TERT mRNA oscillation, and shortened telomere length. Physicians with regular work schedules have circadian oscillation of telomerase activity while emergency physicians working in shifts lose the circadian rhythms of telomerase activity. These findings identify the circadian rhythm as a mechanism underlying telomere and telomerase activity control that serve as interconnections between circadian systems and aging.” (4)
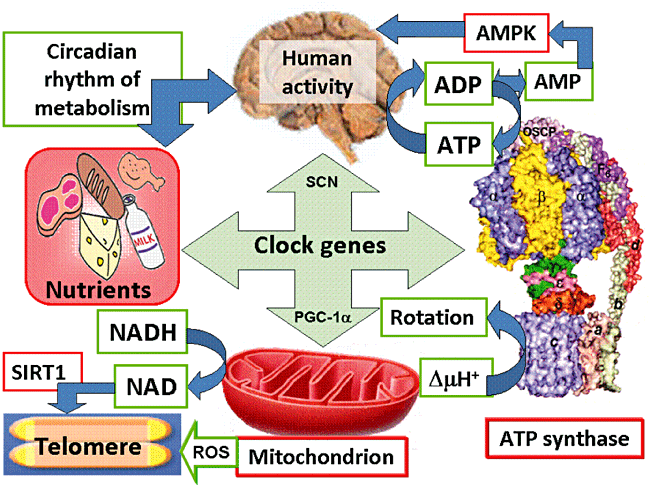
“Human activity is driven by NADH and ATP produced from nutrients, and the resulting NAD and AMP play a predominant role in energy regulation. Caloric restriction increases both AMP and NAD and is known to extend the healthspan (healthy lifespan) of animals. Silent information regulator T1 (SIRT1), the NAD-dependent deacetylase, attenuates telomere shortening, while peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α), a master modulator of gene expression, is phosphorylated by AMP kinase and deacetylated by SIRT1. Thus, PGC-1α is a key component of the circadian oscillator that integrates the mammalian clock and energy metabolism.
Reactive oxygen species produced in clock mutants result in telomere shortening. The circadian rhythms produced by clock genes and lifestyle factors are ultimately controlled by the human brain and drive homeostatic and hedonic feeding and daily activity.” (9)
Short Telomeres in the Heart Associated with Heart Weight Gain and Cause of Death.
“The impairment in cell division, the enhanced cardiac myocyte death and cellular hypertrophy, are concomitant with ventricular dilation, thinning of the wall and cardiac dysfunction. Thus, inhibition of cardiac myocyte replication provoked by telomere shortening, results in de-compensated eccentric hypertrophy and heart failure in mice. Telomere shortening with age could also contribute to cardiac failure in humans, opening the possibility for new therapies.” (1)

“Telomeres are tandem repeat DNA sequences present at the ends of each eukaryotic chromosome to stabilize the genome structure integrity. Telomere lengths progressively shorten with each cell division. Inflammation and oxidative stress, which are implicated as major mechanisms underlying cardiovascular diseases, increase the rate of telomere shortening and lead to cellular senescence.
In clinical studies, cardiovascular risk factors such as smoking, obesity, sedentary lifestyle, and hypertension have been associated with short leukocyte telomere length. In addition, low telomerase activity and short leukocyte telomere length have been observed in atherosclerotic plaque and associated with plaque instability, thus stroke or acute myocardial infarction. The aging myocardium with telomere shortening and accumulation of senescent cells limits the tissue regenerative capacity, contributing to systolic or diastolic heart failure. In addition, patients with ion-channel defects might have genetic imbalance caused by oxidative stress-related accelerated telomere shortening, which may subsequently cause sudden cardiac death. Telomere length can serve as a marker for the biological status of previous cell divisions and DNA damage with inflammation and oxidative stress.” (10)

Telomerase Directing Mesenchymal Stem Cells Toward Cardiac Regeneration.
“Our study shows that hTERT can activate pro-regenerative signalling within PDGFRα + cMSCs and enhance cardiac repair after myocardial infarction… First, AAV9-Tert gene therapy preferentially results in increased Tert expression in cardiomyocytes rather than in progenitor cells and fibroblasts.” (5)
“Myofibroblast differentiation, which depends on complex I activity, was abrogated in TERT-deficient and nucTERT cardiac fibroblasts and completely restored in mitoTERT cells.” (7)
“Mitochondrial TERT protects cardiomyocytes from apoptosis, improves cardiac myofibroblast differentiation, and endothelial cell migration and vascularization.” (7)
Telomerase Enzyme Alleviated Alzheimer’s Symptoms Beyond Rebuilding Telomere.
“However, when the researchers introduced an artificial TERT-producing gene to maintain healthy levels of TERT in the brain even as the mice aged and developed AD, this slowed disease progression and accumulation of amyloid beta (Aß), the hallmark of Alzheimer’s. In conditional mutants – mice that had their artificial TERT gene switched on in the middle of AD progression – TERT activation also led to a striking decline in Aß accumulation in the hippocampus.
TERT induction had several other benefits: it alleviated AD-associated neuroinflammation, resulted in decreased expression of amyloid precursor protein (APP), increased neuronal health, and, finally, led to a significant improvement in cognitive function.”
“Interestingly, TERT induction in human neurons not only decreased APP protein levels but also triggered the activation of the well-known anti-aging gene SIRT1 as well as of several genes related to synaptic plasticity and inflammation suppression.”
“The scientists hypothesize that this non-catalytic neuroprotective activity by TERT might be the reason why neurons maintain a certain level of TERT expression despite being post-mitotic (not dividing anymore). The researchers also note that, according to previous studies, TERT reactivation improves function of other postmitotic cells such as cardiomyocytes and hepatocytes – apparently, without telomere replenishment being involved.” (6)
Telomere Shortening in Neural Stem Cells Disrupts Neuronal Differentiation and Neuritogenesis.
“The negative effect of p53 on neuritogenesis is mechanistically linked to its cooperation with the Notch pathway in the upregulation of small GTPase RhoA kinases, Rock1 and Rock2, suggesting a potential link between DNA damage and the Notch signaling pathway in the control of neuritogenesis. We also show that telomerase expression is downregulated in the SEZ of aging mice leading to telomere length reductions in neurosphere-forming cells and deficient neurogenesis and neuritogenesis. Our results suggest that age-related deficits could be caused partly by dysfunctional telomeres and demonstrate that p53 is a central modulator of adult neurogenesis, regulating both the production and differentiation of postnatally generated olfactory neurons.” (8)

“Removal of p53 is sufficient to rescue defects in proliferation, self-renewal, and differentiation in the SEZ of telomerase-deficient mice… All deleterious effects of critically short telomeres on adult neurogenesis, including those on neuronal differentiation, are mediated by p53.” (8)

“Neurite sprouting is ultimately dependent on pathways regulating actin dynamics and an inverse relationship has been described between the induction of the small GTPase actin-regulator RhoA or its downstream effectors, the RhoA-associated coiled serine/threonine kinases (Rock1 and Rock2), and neurite extension. Cooperative positive effects of p53 and Notch in regulating the expression of Rock1 and 2 have been reported in keratinocytes. Because our data revealed a putative interplay between Notch signaling and p53, we decided to analyze their interaction in the regulation of these kinases. Both Rock1 and Rock2 mRNA levels were under the control of p53 and Notch in our cultures. We observed reduced levels of Rock1 and 2 in adult cells lacking p53 and increased levels in G5 Terc-deficient fetal cultures that have higher levels of p53. NotchIC overexpression also resulted in increased mRNA levels for both genes, and more importantly, it rescued the reduction observed in p53−/− cells to wild-type levels (Fig. 7A). These data suggested that p53 and Notch could act in concert to regulate Rock1 and 2 transcriptions in neural cells as they do in keratinocytes.”
“It is becoming increasingly clear that adult stem cells do bear an unlimited capacity to replicate themselves, partly because telomerase activity may not be sufficient to completely preserve telomere lengths in lifelong replicating cells. A relationship between telomere dysfunction in stem cells and aging is supported by the accelerated senescent phenotype of telomerase-deficient mice. The observations that NSC telomeres shorten with age and that neurogenesis is impaired in telomerase-deficient mice suggest that telomere dynamics may also play a role in neurogenic niches during aging.
Our results support a model whereby accumulated telomere shortening do impact on the potential of NSC populations to persist but also on the capacity of their progeny to terminal differentiate into fully mature neurons, suggesting that age-associated physiological impairments in neural structures that are supported by brainstem cells may additionally be attributable to abnormal differentiation. Our results also indicate that p53 is a central regulator of a novel checkpoint that limits the ability of different somatic stem cells with dysfunctional telomeres to regenerate adult tissues. In addition, the observation that the proliferative advantage displayed by p53-deficient cells can be counteracted by telomere shortening supports the notion that absence of telomere maintenance in postnatal individuals may act as a tumor-suppressive mechanism.”
Sourcing Studies:
6. “Telomerase Alleviates Alzheimer’s Symptoms Not via Telomeres.” Www.lifespan.io, www.lifespan.io/news/telomerase-alleviates-alzheimers-symptoms-not-via-telomeres/.